After getting started and getting acquainted with DNA sequencing data, it's finally time to explore DNA variation. A tool that makes this easy is GEMINI and this post briefly demonstrates some of its functionality. I have only used GEMINI sparingly and what I know about the tool is gathered mostly from their documentation and tutorials. Be sure to check them out if you're planning on using GEMINI.
But before we begin, I'd like to mention that I recently found out that my affiliation with the University of Western Australia entitled me to some free cloud computing. It's not much but now I can stop using my AWS EC2 instance, which will stop being free this coming May. I bring this up because most of the analyses below (except the ESP6500 analysis) were performed using the free cloud space on a new Ubuntu instance. As mentioned in the documentation, several dependencies need to be installed on minimalistic instances. Anyway, let's get a move on.
Installing GEMINI using the automated script
I've installed GEMINI on my Mac and on various Linux distributions, and each time the automated installation worked well. Otherwise you can manually install GEMINI.
# installing on Ubuntu cat /etc/lsb-release DISTRIB_ID=Ubuntu DISTRIB_RELEASE=14.04 DISTRIB_CODENAME=trusty DISTRIB_DESCRIPTION="Ubuntu 14.04.2 LTS" # create a new directory for GEMINI mkdir gemini wget https://raw.github.com/arq5x/gemini/master/gemini/scripts/gemini_install.py python gemini_install.py . gemini # the installation adds the binaries to ~/bin # make sure that is in your $PATH gemini --version gemini 0.18.0
You can also download the GERP and CADD scores for each base of the human genome.
# if you're interested, here's how the data was compressed # check out https://gist.github.com/arq5x/9378020 # the gist will also help you understand # what the columns in the file represent # ~35G gemini update --dataonly --extra cadd_score # ~7G gemini update --dataonly --extra gerp_bp
Installing vt
The vt program is used to decompose and normalise VCF files. I wrote about these procedures in my previous post. Installing the program is straightforward.
# I had to install git on my instance # sudo apt-get install git git clone https://github.com/atks/vt.git cd vt make make test
Installing Perl and VEP
The Variant Effect Predictor program is written in Perl and if you're working on a server without root privileges, I recommend ActivePerl; it makes it easier to install packages. (Since I used a cloud instance, I had root privileges and could make global installations using the CPAN module.) After ActivePerl is installed and added to your $PATH, install these packages using the ActivePerl package manager:
# when you type `which perl` make sure # it points to the ActivePerl installation ppm install Archive::Extract ppm install DBD::mysql
For VEP, I downloaded the cache data manually and extracted it into the .vep directory. The cache data can be downloaded during the installation step when running the INSTALL.pl script but I did it in a separate step.
# the cache data is ~4.5G mkdir ~/.vep cd ~/.vep/ wget -c ftp://ftp.ensembl.org/pub/release-82/variation/VEP/homo_sapiens_vep_82_GRCh37.tar.gz tar -xzf homo_sapiens_vep_82_GRCh37.tar.gz cd wget https://github.com/Ensembl/ensembl-tools/archive/release/82.zip unzip 82.zip cd ensembl-tools-release-82/scripts/variant_effect_predictor perl INSTALL.pl
There is a example VCF file that you can use, to test the installation:
variant_effect_predictor.pl -i example_GRCh37.vcf --cache --assembly GRCh37 --offline --force_overwrite
Downloading test data
In The Exomiser (version 6.0.0) data directory is a VCF file that contains a pathogenic variant for Pfeiffer syndrome. I've hosted this file on my server (~8M) but if you want to download The Exomiser as well, follow the steps below. Otherwise, just download the file from my server.
# download distribution ~1.3G wget --continue ftp://ftp.sanger.ac.uk/pub/resources/software/exomiser/downloads/exomiser/old_versions/6.0.0/exomiser-cli-6.0.0-distribution.zip unzip exomiser-cli-6.0.0-distribution.zip # checksum md5sum exomiser-cli-6.0.0/data/Pfeiffer.vcf bf81821b18a6c91ef821a30c42aebc66 Pfeiffer.vcf
Downloading the reference genome
The vt normalise step requires a reference genome. If you look inside Pfeiffer.vcf, you'll notice that the chromosome names are 1, 2, 3, etc. If you're a UCSC Genome Browser fan, like myself, you're probably used to the chr1, chr2, chr3, etc. convention; the 1, 2, 3, etc. convention is used by the 1000 Genomes project. There's an excellent
article that explains the conventions and the differences between the 1000 Genomes and UCSC Genome Browser references. As I found out, this difference in naming, causes some annoyance.
wget -c ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/technical/reference/human_g1k_v37.fasta.gz gunzip human_g1k_v37.fasta.gz # to save space and to allow # tabix indexing bgzip human_g1k_v37.fasta
GEMINI workflow
Now that we have everything in place, we can go through the GEMINI workflow. First, we decompose and normalise our VCF file (Pfeiffer.vcf). Then we annotate the variants using a variant annotation tool; I have decided to go with VEP. And lastly, we use GEMINI to create a sqlite database (combining other data sources as well) from the VCF file.
# First decompose and normalise vt decompose -s Pfeiffer.vcf | vt normalize -r human_g1k_v37.fasta.gz - > Pfeiffer_vt.vcf # Annotate using VEP perl ~/work/ensembl-tools-release-82/scripts/variant_effect_predictor/variant_effect_predictor.pl -i Pfeiffer_vt.vcf -o Pfeiffer_vt_vep.vcf --vcf \ --offline --cache --sift b --polyphen b --symbol --numbers --biotype --total_length \ --fields Consequence,Codons,Amino_acids,Gene,SYMBOL,Feature,EXON,PolyPhen,SIFT,Protein_position,BIOTYPE # finally load into GEMINI gemini load -v Pfeiffer_vt_vep.vcf -t VEP Pfeiffer_vt_vep.db
Exploring the variants
This excellent tutorial explains the basics of making queries using GEMINI. I also wrote a blog post on SQLite. To know what tables and columns are available, check out the GEMINI database schema. Below are some basic queries I came up with.
# how many variants?
gemini query -q 'select count(*) from variants' --header Pfeiffer_vt_vep.db
count(*)
37652
# how many SNPs and INDELs?
gemini query -q 'select type,count(*) from variants group by type' --header Pfeiffer_vt_vep.db
type count(*)
indel 3844
snp 33808
# core VCF fields
gemini query -q 'select chrom,start,end,vcf_id,variant_id,anno_id,ref,alt,qual,filter from variants limit 10' --header Pfeiffer_vt_vep.db
chrom start end vcf_id variant_id anno_id ref alt qual filter
chr1 866510 866511 rs60722469 1 1 C CCCCT 258.619995117 None
chr1 879316 879317 rs7523549 2 2 C T 150.770004272 None
chr1 879481 879482 None 3 3 G C 484.519989014 None
chr1 880389 880390 rs3748593 4 11 C A 288.440002441 None
chr1 881626 881627 rs2272757 5 4 G A 486.239990234 None
chr1 884090 884091 rs7522415 6 4 C G 65.4599990845 None
chr1 884100 884101 rs4970455 7 2 A C 85.8099975586 None
chr1 892459 892460 rs41285802 8 6 G C 1736.69995117 None
chr1 897729 897730 rs7549631 9 5 C T 225.339996338 None
chr1 909237 909238 rs3829740 10 3 G C 247.539993286 None
# the impact severity of the variants
gemini query -q 'select impact_severity,count(*) from variants group by impact_severity' --header Pfeiffer_vt_vep.db
impact_severity count(*)
HIGH 256
LOW 31930
MED 5466
# how many variants are in ClinVar
# and what are their annotation/s
gemini query -q 'select clinvar_sig,count(*) from variants group by clinvar_sig' --header Pfeiffer_vt_vep.db
clinvar_sig count(*)
None 36443
10
benign 639
benign,drug-response,other 1
benign,other 17
benign,pathogenic 3
benign,pathogenic,other 1
drug-response 7
drug-response,other 1
likely-benign 231
likely-benign,benign 41
likely-benign,benign,other 1
likely-benign,other 3
not-provided 65
not-provided,benign 13
not-provided,benign,other 2
not-provided,likely-benign 1
not-provided,other 6
not-provided,pathogenic 4
not-provided,pathogenic,other 1
not-provided,uncertain,benign,other 1
other 121
other,drug-response 1
pathogenic 11
pathogenic,other 7
uncertain 15
uncertain,benign 5
uncertain,likely-benign,benign 1
# find variants that are listed as
# pathogenic in ClinVar
gemini query -q 'select chrom,start,end,ref,alt,qual,gene,clinvar_disease_name from variants where clinvar_sig like "%pathogenic%"' --header Pfeiffer_vt_vep.db
chrom start end ref alt qual gene clinvar_disease_name
chr1 70904799 70904800 G T 859.229980469 CTH Homocysteine,_total_plasma,_elevated
chr1 94512564 94512565 C T 1556.08996582 ABCA4 MACULAR_DEGENERATION,_AGE-RELATED,_2,_SUSCEPTIBILITY_TO|Stargardt_disease_1|not_provided|not_specified
chr1 98348884 98348885 G A 830.869995117 DPYD Dihydropyrimidine_dehydrogenase_deficiency,not_provided
chr1 223285199 223285200 G A 486.880004883 TLR5 Legionellosis|Systemic_lupus_erythematosus,_resistance_to,_1|Melioidosis,_resistance_to
chr1 231408090 231408091 A G 1555.84997559 GNPAT Rhizomelic_chondrodysplasia_punctata_type_2
chr2 233243585 233243586 C T 381.769989014 ALPP ALKALINE_PHOSPHATASE,_PLACENTAL,_ALLELE-1_POLYMORPHISM|ALPP*1,ALKALINE_PHOSPHATASE,_PLACENTAL,_ALLELE-3_POLYMORPHISM|ALPP*3
chr3 10331456 10331457 G T 1153.54003906 GHRL Obesity,_age_at_onset_of|Metabolic_syndrome,_susceptibility_to
chr3 38645419 38645420 T C 129.509994507 SCN5A Progressive_familial_heart_block_type_1A|not_specified|not_provided
chr3 39307255 39307256 C T 897.16998291 CX3CR1 Human_immunodeficiency_virus_type_1,_rapid_progression_to_AIDS|Coronary_artery_disease,_resistance_to|Age-related_macular_degeneration_12|MACULAR_DEGENERATION,_AGE-RELATED,_12,_SUSCEPTIBILITY_TO
chr3 165491279 165491280 C T 173.919998169 BCHE BCHE,_K_variant|CHE*539T
chr4 187004073 187004074 C T 175.009994507 TLR3 Congenital_human_immunodeficiency_virus
chr5 35861067 35861068 T C 989.719970703 IL7R Severe_combined_immunodeficiency,_autosomal_recessive,_T_cell-negative,_B_cell-positive,_NK_cell-positive|not_specified
chr5 35871189 35871190 G A 400.589996338 IL7R Severe_combined_immunodeficiency,_autosomal_recessive,_T_cell-negative,_B_cell-positive,_NK_cell-positive|not_specified
chr5 149212242 149212243 G C 2727.2800293 PPARGC1B Obesity,_variation_in
chr7 94946083 94946084 A T 906.369995117 PON1 Coronary_artery_disease,_susceptibility_to|Microvascular_complications_of_diabetes_5|Enzyme_activity_finding
chr9 132580900 132580901 C G 657.179992676 TOR1A Dystonia_1,_torsion,_modifier_of|Dystonia_1
chr10 101829513 101829514 C T 421.380004883 CPN1 Anaphylotoxin_inactivator_deficiency
chr11 68855362 68855363 G A 59.3199996948 TPCN2 Skin/hair/eye_pigmentation,_variation_in,_10
chr11 89017960 89017961 G A 258.910003662 TYR Oculocutaneous_albinism_type_1B|Oculocutaneous_albinism_type_1,_temperature_sensitive|Cutaneous_malignant_melanoma_8|Skin/hair/eye_pigmentation,_variation_in,_3|Skin/hair/eye_pigmentation_3,_blue/green_eyes|WAARDENBURG_SYNDROME_2_AND_OCULAR_ALBINISM,_DIGENIC|not_provided
chr11 111635565 111635566 C T 335.880004883 PPP2R1B Lung_cancer
chr12 6458349 6458350 A G 1335.06994629 SCNN1A Bronchiectasis_with_or_without_elevated_sweat_chloride_2
chr14 21790039 21790040 G T 97.0100021362 RPGRIP1 Cone-rod_dystrophy_13|not_provided
chr17 12915008 12915009 G A 650.349975586 ELAC2 Prostate_cancer,_hereditary,_2
chr22 18905963 18905964 C T 164.479995728 PRODH Proline_dehydrogenase_deficiency|Schizophrenia_4
chr22 42523942 42523943 A G 530.0 CYP2D6 Debrisoquine,_ultrarapid_metabolism_of
chr22 51064038 51064039 G C 650.539978027 ARSA Metachromatic_leukodystrophy|not_specified
chr10 123256214 123256215 T G 100.0 FGFR2 Pfeiffer_syndrome
The last query, outputs variants that are in the ClinVar database and listed as pathogenic. On the very last line of the output is the causative variant for Pfeiffer syndrome (unintentionally convenient), which is located in the FGFR2 gene. (This is also the variant that The Exomiser prioritises, along with two other intronic variants in the FGFR2 gene.) In addition, this variant is not present in HapMap, ESP, the 1000 Genomes project, and ExAC.
gemini query -q 'select in_hm2,in_hm3,in_esp,in_1kg,in_exac from variants where clinvar_disease_name == "Pfeiffer_syndrome"' --header Pfeiffer_vt_vep.db in_hm2 in_hm3 in_esp in_1kg in_exac None None 0 0 0
What are the PolyPhen, SIFT, and CADD scores of this variant?
gemini query -q 'select polyphen_score,sift_score,cadd_scaled from variants where clinvar_disease_name == "Pfeiffer_syndrome"' --header Pfeiffer_vt_vep.db polyphen_score sift_score cadd_scaled 0.907 0.02 23.7
How do these scores compare to the rest of the variants in the VCF file?
gemini query -q 'select polyphen_score,sift_score,cadd_scaled from variants' --header Pfeiffer_vt_vep.db > score.tsv
Load and plot using R:
df <- read.table('score.tsv', header=TRUE, stringsAsFactors = FALSE)
df$polyphen_score <- gsub(pattern = "None", replacement = NA, x = df$polyphen_score)
df$sift_score <- gsub(pattern = "None", replacement = NA, x = df$sift_score)
df$cadd_scaled <- gsub(pattern = "None", replacement = NA, x = df$cadd_scaled)
par(mfrow = c(3,1))
plot(density(as.numeric(na.omit(df$polyphen_score))), main="PolyPhen scores")
plot(density(as.numeric(na.omit(df$sift_score))), main="SIFT scores")
plot(density(as.numeric(na.omit(df$cadd_scaled))), main="CADD scaled scores")
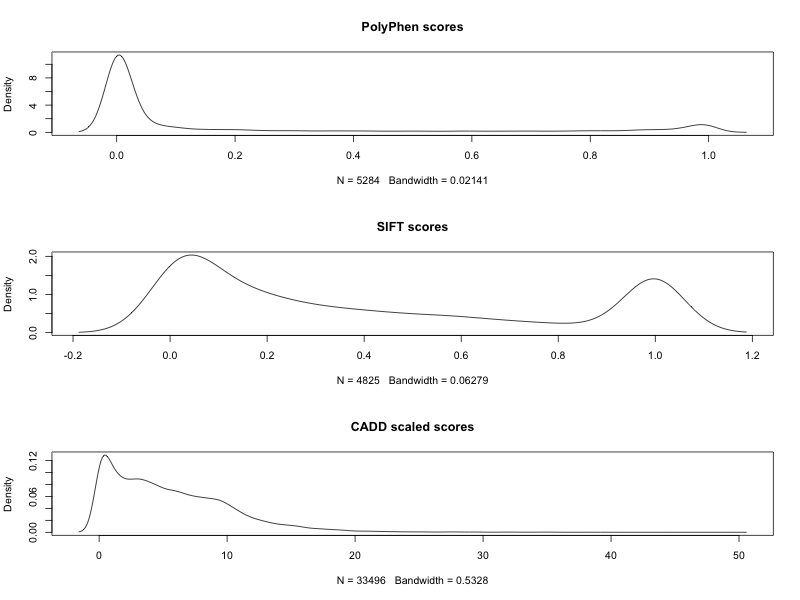 The higher the PolyPhen score, the higher the impact; the lower the SIFT probability score, the higher the impact; and the higher the CADD score, the higher the likelihood of the SNV having a damaging effect.
The higher the PolyPhen score, the higher the impact; the lower the SIFT probability score, the higher the impact; and the higher the CADD score, the higher the likelihood of the SNV having a damaging effect.
However, I'm not sure where the other variants in the Pfeiffer.vcf file came from (and whether they are also pathogenic or not). The next section uses GEMINI to explore variants in the NHLBI Grand Opportunity Exome Sequencing Project.
ESP6500
The NHLBI Grand Opportunity Exome Sequencing Project (ESP) provides a list of variants called from 15 separate cohorts that includes over 2,000 exomes. The supplementary data in Tennessen et al., 2012 describes the cohorts and the disease phenotypes associated with the exomes. The variants can be downloaded from the Exome Variant Server; the latest version is ESP6500. For this part of the post, I want to annotate and process the variants in the ESP6500 dataset.
# download and extract # I realise it says GRCh38-liftover # but these variants are on GRCh37 coordinates, I checked wget -c http://evs.gs.washington.edu/evs_bulk_data/ESP6500SI-V2-SSA137.GRCh38-liftover.snps_indels.vcf.tar.gz tar -xzf ESP6500SI-V2-SSA137.GRCh38-liftover.snps_indels.vcf.tar.gz # the VCF files are split by chromosome # the steps below stitch them back together # bgzip and index parallel bgzip ::: *.vcf parallel tabix -p vcf ::: *.vcf.gz # merge all the VCF files bcftools merge -o ESP6500SI-V2-SSA137.all.vcf.gz -O z *.vcf.gz # there's probably a better way # to sort a file with comments gunzip -c ESP6500SI-V2-SSA137.all.vcf.gz | grep '^#' > out.vcf gunzip -c ESP6500SI-V2-SSA137.all.vcf.gz | grep -v '^#' | sort -k1,1V -k2,2n >> out.vcf bgzip out.vcf mv -f out.vcf.gz ESP6500SI-V2-SSA137.all.vcf.gz # decompose and normalise # I have a hg19.fa file where the "chr" # has been removed vt decompose -s ESP6500SI-V2-SSA137.all.vcf.gz | vt normalize -r hg19_no_chr.fa - | gzip > ESP6500SI-V2-SSA137.all.vt.vcf.gz # annotate using VEP perl ~/src/ensembl-tools-release-82/scripts/variant_effect_predictor/variant_effect_predictor.pl -i ESP6500SI-V2-SSA137.all.vt.vcf.gz -o ESP6500SI-V2-SSA137.all.vt.vep.vcf --vcf \ --offline --cache --sift b --polyphen b --symbol --numbers --biotype --total_length \ --fields Consequence,Codons,Amino_acids,Gene,SYMBOL,Feature,EXON,PolyPhen,SIFT,Protein_position,BIOTYPE # load into gemini # I got a panic attack when I loaded with 64 cores # because our main server (96 cores with 1TB RAM) became unresponsive # However, after 30 or so minutes, the server came back to life (phew!) # I think it was a disk issue gemini load -v ESP6500SI-V2-SSA137.all.vt.vep.vcf -t VEP --cores 64 ESP6500SI-V2-SSA137.all.vt.vep.db
After successfully creating the database using GEMINI, I performed some of the queries as above.
gemini query -q 'select count(*) from variants' --header ESP6500SI-V2-SSA137.all.vt.vep.db count(*) 1998204 gemini query -q 'select type,count(*) from variants group by type' --header ESP6500SI-V2-SSA137.all.vt.vep.db type count(*) indel 125311 snp 1872893 gemini query -q 'select chrom,start,end,vcf_id,variant_id,anno_id,ref,alt,qual,filter from variants limit 10' --header ESP6500SI-V2-SSA137.all.vt.vep.db chrom start end vcf_id variant_id anno_id ref alt qual filter chr1 69427 69428 rs140739101 1 1 T G None None chr1 69475 69476 rs148502021 2 1 T C None None chr1 69495 69496 rs150690004 3 1 G A None None chr1 69510 69511 rs75062661 4 1 A G None None chr1 69589 69590 rs141776804 5 1 T A None None chr1 69593 69594 rs144967600 6 1 T C None None chr1 69619 69621 None 7 1 TA T None None chr1 69760 69761 rs200505207 8 1 A T None None chr1 801942 801943 rs7516866 9 1 C T None None chr1 801975 801976 rs377032113 10 1 C T None None gemini query -q 'select impact_severity,count(*) from variants group by impact_severity' --header ESP6500SI-V2-SSA137.all.vt.vep.db impact_severity count(*) HIGH 51861 LOW 1219612 MED 726731
As reported in Tennessen et al., 2012, over 50% of their variants were predicted to be missense variants, i.e. a single nucleotide variant that causes an amino acid change. Missense variants are often annotated with medium severity and thus the bulk of the variants with medium severity are the missence variants.
gemini query -q 'select impact_severity, impact, count(impact) from variants group by impact' --header ESP6500SI-V2-SSA137.all.vt.vep.db | sort -k3n impact_severity impact count(impact) MED protein_altering_variant 66 LOW mature_miRNA_variant 134 LOW coding_sequence_variant 139 LOW stop_retained_variant 395 HIGH stop_lost 794 HIGH start_lost 1243 MED inframe_insertion 2249 LOW intergenic_variant 3814 HIGH splice_acceptor_variant 3926 LOW downstream_gene_variant 4412 HIGH splice_donor_variant 4660 LOW upstream_gene_variant 6878 MED inframe_deletion 10655 LOW non_coding_transcript_exon_variant 10902 HIGH stop_gained 17528 HIGH frameshift_variant 23710 LOW 5_prime_UTR_variant 34229 LOW 3_prime_UTR_variant 47340 LOW splice_region_variant 71338 LOW synonymous_variant 423787 LOW intron_variant 616244 MED missense_variant 713761
Lastly, let's plot the distribution of PolyPhen, SIFT, and CADD scores for the ESP6500 dataset.
gemini query -q 'select polyphen_score,sift_score,cadd_scaled from variants' --header ESP6500SI-V2-SSA137.all.vt.vep.db > score.tsv
Using the same R code as above, we can create the same plots.
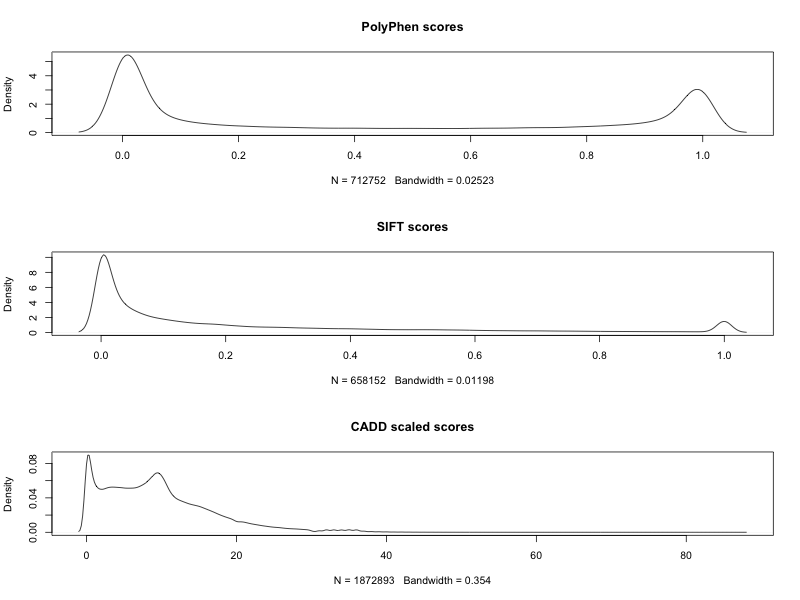 The large number of variants with high impact SIFT scores (low probability scores), are probably the missense variants. There's also a peak at around 10 for the scaled CADD scores that should be interesting to look into.
The large number of variants with high impact SIFT scores (low probability scores), are probably the missense variants. There's also a peak at around 10 for the scaled CADD scores that should be interesting to look into.
Summary
In this post, I showed the steps required to get GEMINI up and running. I used VEP to annotate two VCF files: a VCF file containing a pathogenic variant for Pfeiffer syndrome and a VCF containing the ESP variants, and processed them using GEMINI. I used GEMINI to output some characteristics of the Pfeiffer syndrome variant and to performed some queries on the ESP variants. I was interested in the PolyPhen, SIFT, and CADD scores of the Pfeiffer syndrome variant and how its score compares with variants in the ESP dataset. The CADD score seems to be a good filter for identifying causative variants.
Further reading
Below is a recent talk by Aaron Quinlan on GEMINI


This work is licensed under a Creative Commons
Attribution 4.0 International License.
Amazing website. How to following your page. LoL
Thanks. You could use http://feedly.com/ (or any RSS feed reader of your choice).