From the original paper describing the Gene Set Enrichment Analysis:
The goal of GSEA is to determine whether members of a gene set S tend to occur toward the top (or bottom) of the list L, in which case the gene set is correlated with the phenotypic class distinction.
The analysis can be illustrated with a figure. (We'll get to how it was made later in the post.)

The x-axis shows the ranked list of genes, L, and the vertical bars on the x-axis show the genes that belong to gene set S, which in this case is the "Cell Cycle" set and the y-axis shows the enrichment score. We can see that a bunch of "Cell Cycle" genes are enriched in the top ranking genes, which is what the green line is showing.
fgsea
For this post, we will be using the fgsea package, so install that first.
if (!require("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("fgsea")
If you get the error message maximal number of DLLs reached, you can increase this number by modifying the Renviron file. This file is located in /Library/Frameworks/R.framework/Resources/etc for me; this may be different if you're using another operating system.
# show where home is Sys.getenv()['R_HOME'] R_HOME /Library/Frameworks/R.framework/Resources
Once you've located the file, add this line "R_MAX_NUM_DLLS=153" to the end of it. I got 153 from here.
Vignette
The following section is based on the fgsea tutorial but with my elaborations. The pathways are stored in examplePathways and the ranked gene list in exampleRanks; see my appendix to see how this file was generated and how the genes were ranked.
data(examplePathways) # pathways are stored in a list class(examplePathways) [1] "list" # the example list has 1,457 "pathways" length(examplePathways) [1] 1457 # this is the first pathway, which contains # all the Entrez gene IDs of the Meiotic_Synapsis pathway examplePathways[1] $`1221633_Meiotic_Synapsis` [1] "12189" "13006" "15077" "15078" "15270" "15512" "16905" [8] "16906" "19357" "20842" "20843" "20957" "20962" "21749" [15] "21750" "22196" "23856" "24061" "28113" "50878" "56739" [22] "57321" "64009" "66654" "69386" "71846" "74075" "77053" [29] "94244" "97114" "97122" "97908" "101185" "140557" "223697" [36] "260423" "319148" "319149" "319150" "319151" "319152" "319153" [43] "319154" "319155" "319156" "319157" "319158" "319159" "319160" [50] "319161" "319565" "320332" "320558" "326619" "326620" "360198" [57] "497652" "544973" "625328" "667250" "100041230" "102641229" "102641751" [64] "102642045" data(exampleRanks) # ranks are stored as a numeric vector class(exampleRanks) [1] "numeric" # the names of the vector are the Entrez gene IDs head(exampleRanks) 170942 109711 18124 12775 72148 16010 -63.33703 -49.74779 -43.63878 -41.51889 -33.26039 -32.77626 # the values are the rank metric head(unname(exampleRanks)) [1] -63.33703 -49.74779 -43.63878 -41.51889 -33.26039 -32.77626 # ranks are from lowest to highest tail(unname(exampleRanks)) [1] 47.58235 49.87543 50.25179 50.86532 51.16110 53.28400
Analysis
Now we will carry out the procedure to test for the enrichment of gene sets at the top and bottom of the ranked genes.
data(examplePathways)
data(exampleRanks)
fgseaRes <- fgsea(pathways = examplePathways,
stats = exampleRanks,
minSize=15,
maxSize=500,
nperm=100000)
# see https://github.com/Rdatatable/data.table/wiki
# if you are unfamiliar with the data.table package
class(fgseaRes)
[1] "data.table" "data.frame"
# top 6 enriched pathways
head(fgseaRes[order(pval), ])
pathway pval padj ES NES nMoreExtreme
1: 5990980_Cell_Cycle 1.226723e-05 0.0002701041 0.5388497 2.680900 0
2: 5990979_Cell_Cycle,_Mitotic 1.255556e-05 0.0002701041 0.5594755 2.743471 0
3: 5991210_Signaling_by_Rho_GTPases 1.317141e-05 0.0002701041 0.4238512 2.010545 0
4: 5991454_M_Phase 1.375667e-05 0.0002701041 0.5576247 2.552055 0
5: 5991023_Metabolism_of_carbohydrates 1.391188e-05 0.0002701041 0.4944766 2.240926 0
6: 5991209_RHO_GTPase_Effectors 1.394739e-05 0.0002701041 0.5248796 2.373183 0
size leadingEdge
1: 369 66336,66977,12442,107995,66442,19361,
2: 317 66336,66977,12442,107995,66442,12571,
3: 231 66336,66977,20430,104215,233406,107995,
4: 173 66336,66977,12442,107995,66442,52276,
5: 160 11676,21991,15366,58250,12505,20527,
6: 157 66336,66977,20430,104215,233406,107995,
# number of significant pathways at padj < 0.01
sum(fgseaRes[, padj < 0.01])
[1] 77
# plot the most significantly enriched pathway
plotEnrichment(examplePathways[[head(fgseaRes[order(pval), ], 1)$pathway]],
exampleRanks) + labs(title=head(fgseaRes[order(pval), ], 1)$pathway)
Plot the top 10 pathways enriched at the top and bottom of the ranked list, respectively.
topPathwaysUp <- fgseaRes[ES > 0][head(order(pval), n=10), pathway]
topPathwaysDown <- fgseaRes[ES < 0][head(order(pval), n=10), pathway]
topPathways <- c(topPathwaysUp, rev(topPathwaysDown))
plotGseaTable(examplePathways[topPathways], exampleRanks, fgseaRes,
gseaParam = 0.5)
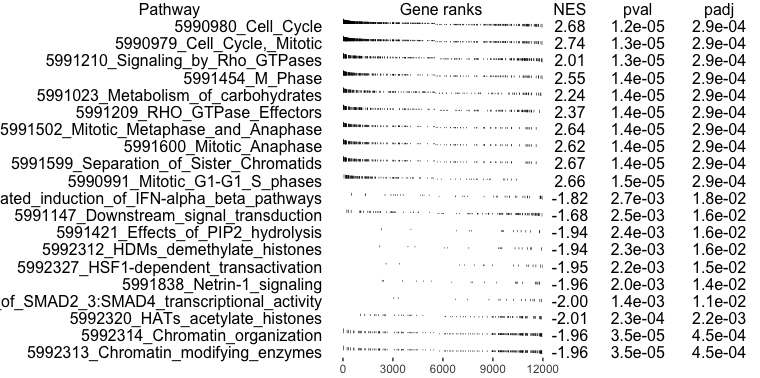
Since the genes were ranked according to their differential expression significance and fold change, with the most significantly down-regulated genes at the top and up-regulated genes at the bottom of the list, the enriched gene sets provides us with some idea of the function of these genes. For example, exampleRanks was generated by testing regulatory T cells against T helper cells, so the enriched pathways may suggest differences between these two types of cells.
Reactome
You can also use Reactome pathways with fgsea. Make sure you have the database installed.
## try http:// if https:// URLs are not supported
source("https://bioconductor.org/biocLite.R")
biocLite("reactome.db")
We'll perform the same analysis as before but this time we'll use the pathways from the Reactome database.
my_pathways <- reactomePathways(names(exampleRanks))
# Reactome pathways have a median of 11 genes
summary(sapply(my_pathways, length))
Min. 1st Qu. Median Mean 3rd Qu. Max.
1.0 4.0 11.0 30.3 30.0 1140.0
fgsea_reactome <- fgsea(pathways = my_pathways,
stats = exampleRanks,
minSize=15,
maxSize=500,
nperm=100000)
head(fgsea_reactome[order(pval), ])
pathway pval padj ES NES nMoreExtreme size
1: Cell Cycle 1.215983e-05 0.0002401877 0.5469310 2.735403 0 393
2: Cell Cycle, Mitotic 1.241111e-05 0.0002401877 0.5570318 2.754600 0 346
3: Neutrophil degranulation 1.264302e-05 0.0002401877 0.4305249 2.103791 0 306
4: Signaling by Rho GTPases 1.303169e-05 0.0002401877 0.4151348 1.985009 0 248
5: M Phase 1.307497e-05 0.0002401877 0.5080161 2.422413 0 242
6: Cell Cycle Checkpoints 1.344737e-05 0.0002401877 0.6158766 2.872592 0 200
leadingEdge
1: 66336,66977,15366,12442,107995,66442,
2: 66336,66977,15366,12442,107995,66442,
3: 11676,14190,53381,12306,20430,12505,
4: 66336,66977,20430,104215,233406,107995,
5: 66336,66977,12442,107995,66442,52276,
6: 66336,66977,12442,107995,66442,12428,
sum(fgsea_reactome[, padj < 0.01])
[1] 105
Leading edge
Again from the original paper:
Often it is useful to extract the core members of high scoring gene sets that contribute to the enrichment score. We define the leading-edge subset to be those genes in the gene set S that appear in the ranked list L at, or before, the point where the running sum reaches its maximum deviation from zero.
As you may have noticed from the previous section, one of the results column was named "leadingEdge"; this contains the genes that contributed to the enrichment score. I'll use the GSEA performed using the Reactome pathways to illustrate.
# the most significant pathway
fgsea_reactome[order(pval),][1,]
pathway pval padj ES NES nMoreExtreme size
1: Cell Cycle 1.215983e-05 0.0002401877 0.546931 2.735403 0 393
leadingEdge
1: 66336,66977,15366,12442,107995,66442,
# list of Entrez gene IDs that contributed to the enrichment score
fgsea_reactome[order(pval),][1,]$leadingEdge
[[1]]
[1] "66336" "66977" "15366" "12442" "107995" "66442" "12571" "12428" "52276" "54392"
[11] "66311" "215387" "67629" "54124" "12649" "72415" "56150" "57441" "20877" "67121"
[21] "12615" "11799" "66468" "67849" "19053" "73804" "76044" "20878" "15270" "13555"
[31] "60411" "12580" "17219" "69270" "12575" "69263" "12448" "14211" "20873" "18005"
[41] "72119" "71988" "12189" "17215" "12534" "66156" "208628" "237911" "22390" "68240"
[51] "228421" "68014" "269582" "19348" "12236" "72151" "18817" "21781" "18968" "217653"
[61] "66934" "272551" "227613" "67141" "68612" "68298" "108000" "23834" "106344" "56452"
[71] "242705" "18141" "223921" "26886" "13557" "26909" "72787" "268697" "72155" "56371"
[81] "17535" "107823" "12531" "327762" "12567" "229841" "67052" "16319" "66634" "26931"
[91] "67203" "12235" "19891" "74470" "72083" "381318" "66570" "17216" "19687" "17218"
[101] "102920" "29870" "18973" "16881" "17463" "75786" "19645" "19075" "26417" "69736"
[111] "19357" "76816" "70385" "70645" "22628" "225182" "22627" "52683" "19076" "18972"
[121] "231863" "26932" "12544" "17997" "26440" "68549" "19088" "269113" "26444" "19324"
[131] "103733" "59001" "107976" "19179" "12579" "232987" "17420" "228769" "219072" "26445"
[141] "105988" "69745" "18538" "69928" "11651" "235559" "68097" "57296" "14235" "19170"
[151] "17246" "17220" "12144" "50793" "18392" "236930" "67151" "70024" "59126" "66296"
[161] "109145" "71819" "67733" "50883" "12447" "75430" "12532" "14156" "26442" "19177"
[171] "230376" "245688"
# how many genes are in the leading edge?
length(fgsea_reactome[order(pval),][1,]$leadingEdge[[1]])
[1] 172
# how many genes are in the Cell Cycle pathway?
length(my_pathways[['Cell Cycle']])
[1] 393
Summary
The Gene Set Enrichment Analysis (GSEA) has been around since 2005 and has become a routine analysis step in gene expression analyses. It differs from Gene Ontology enrichment analysis in that it considers all genes in contrast to taking only significantly differentially expressed genes. The fgsea package allows one to conduct a pre-ranked GSEA in R, which is one approach in a GSEA. A p-value is estimated by permuting the genes in a gene set, which leads to randomly assigned gene sets of the same size. Note that "This approach is not strictly accurate because it ignores gene-gene correlations and will overestimate the significance levels and may lead to false positives." However, it is still useful in getting an idea of the potential roles of the genes that are up- and down-regulated. If your pre-ranked GSEA returns no significant gene sets, you may still get an idea of what roles the up- and down-regulated genes may be involved in by examining the leading edge set. This set indicates the genes that contributed to the enrichment score.
The example ranks in the fgsea package were ranked on the moderated t-statistic. Another metric that you may consider is to simply take the signed fold change and multiply it by the -log10p-value. For example, if you performed your differential expression analysis with edgeR, you can simply multiply the signed fold change column to the -log10p-value column. This metric is based on this paper: Rank-rank hypergeometric overlap: identification of statistically significant overlap between gene-expression signatures.
Appendix
The author of fgsea provides the R script for generating exampleRanks; for convenience the code is provided below.
source("https://raw.githubusercontent.com/assaron/r-utils/master/R/exprs.R")
gse14308 <- getGEO("GSE14308")[[1]]
pData(gse14308)$condition <- sub("-.*$", "", gse14308$title)
es <- collapseBy(gse14308, fData(gse14308)$ENTREZ_GENE_ID, FUN=median)
es <- es[!grepl("///", rownames(es)), ]
es <- es[rownames(es) != "", ]
exprs(es) <- normalizeBetweenArrays(log2(exprs(es)+1), method="quantile")
es.design <- model.matrix(~0+condition, data=pData(es))
fit <- lmFit(es, es.design)
fit2 <- contrasts.fit(fit, makeContrasts(conditionTh1-conditionNaive,
levels=es.design))
fit2 <- eBayes(fit2)
de <- data.table(topTable(fit2, adjust.method="BH", number=12000, sort.by = "B"), keep.rownames = T)
ranks <- de[order(t), list(rn, t)]
The genes are ranked by the moderated t-statistic, where the sign indicates the fold change
de[order(t), c(1,19:24)]
rn logFC AveExpr t P.Value adj.P.Val B
1: 170942 -6.400779 11.670183 -62.22877 5.412495e-11 8.621514e-07 13.91586
2: 109711 -5.655664 11.107294 -49.47829 2.752658e-10 8.621514e-07 13.15661
3: 18124 -5.829724 8.730385 -43.40540 6.963327e-10 1.119183e-06 12.62986
4: 12775 -5.066777 11.472139 -41.16952 1.012754e-09 1.314681e-06 12.39775
5: 72148 -4.864065 8.365239 -33.23463 4.606844e-09 3.417291e-06 11.34801
---
11996: 58801 6.730286 9.721764 49.10222 2.905662e-10 8.621514e-07 13.12780
11997: 13730 8.979133 8.379178 50.02863 2.545022e-10 8.621514e-07 13.19797
11998: 15937 5.519076 9.440537 50.29120 2.452293e-10 8.621514e-07 13.21737
11999: 12772 8.833193 9.721834 50.52651 2.372453e-10 8.621514e-07 13.23458
12000: 80876 6.897013 12.040680 52.59930 1.783927e-10 8.621514e-07 13.37922
Visualise the six most significantly down- and up-regulated genes.
library(pheatmap) my_group <- data.frame(group = pData(es)$condition) row.names(my_group) <- colnames(exprs(es)) pheatmap( mat = es[c(head(de[order(t), 1])$rn, tail(de[order(t), 1])$rn),], annotation_col = my_group, cluster_rows = FALSE, cellwidth=25, cellheight=15 )



This work is licensed under a Creative Commons
Attribution 4.0 International License.
Hello,
I am a graduate student. I have pre-ranked list of genes, but it is not a comprehensive gene list. How can I find a reference/background set that would complete my list? My gene set is from brain cortex and it compares normal vs alzheimer’s disease samples age >65.
I really appreciate the help
I forgot to mention the samples come from post-mortem human tissue.
Hello,
how were the pre-ranked list of genes compiled in the first place? You will need carry out some sort of differential gene expression analysis between normal and disease samples and save the fold changes and p-values for all the genes that were tested.
Best,
Dave
I am sorry I should have explained myself a little more. That is the thing. I was handed this data as is like that. it had a list of genes with p-value and fold change.
I assume these were results of differential expression. But I know it wouldn’t be the whole list of genes detected because there was some kind of cutoff to produce downregulated and upregulated gene lists. I want to perform pathway enrichment analysis and gene set enrichment analysis but I would like a good reference or background gene set to add to the gene lists when doing the enrichment analysis. I am having trouble finding an article or a resource to help me with this. I wondered about using RNA-seq data from the brain atlas but I wasn’t sure how accurate or valid that would be for my study.
Your help is greatly appreciated and thank you for responding,
Vivian
Do you have access to the raw data? You might be better off (in the long run) to redo the differential analysis; this way you’ll have all the p-values and fold changes, and you can select an appropriate background for a GO enrichment analysis. You could use some brain atlas to help you select a more suitable background for a GO enrichment analysis but as far as I’m aware, you can’t perform a GSEA without the full list of genes that were assayed.
Thanks.
How do I go about getting a background gene set for the brain though? for over-representation analysis for example.
Where can I find a brain genome that I can reference when doing an analysis and don’t want to include the whole genome.
Try searching through GEO to find a suitable dataset https://www.ncbi.nlm.nih.gov/gds/?term=human%20brain%20cortex. Then use the genes that are expressed in the tissue of interest as your background.
Dear Davo,
From my understanding FGSEA was implemented by Korotkevich.
https://www.biorxiv.org/content/10.1101/060012v2.full.
However you cite Subramanian af the author? I am a bit confused is both refering to the same article?
Hi,
the GSEA method was developed by Subramanian et al. FGSEA is an implementation of GSEA and was developed by Korotkevich et al.
Dave
Hi Davo,
I had the same problem on heatplot: https://github.com/YuLab-SMU/clusterProfiler.dplyr/issues/2.
Did you solve it? Would you mind share it?
Best
Hi there,
I didn’t figure it out. I just noticed that the repository had been archived. Looks like it won’t be addressed.
Hi Davo,
Thanks for the tutorial. I was trying to find a comprehensive tutorial which discusses the pre-ranking of the gene list and this is it. Just one slight issue. I am using a DE result table from DESeq2. Initially, I used the stat column from the table as rank metric and for a certain gene list. Now, after reading this tutorial I used log2FoldChange*-log10(p) as rank metric. Unfortunately, the ES and NES are in completley opposite directions. Stat metric shows a negative enrichment while the new metric shows a positive enrichment. Any guesses why? I really appreciate your help.
Best wishes
Nurun
Hello Davo,
I am currently doing a project for one of my bioinformatics classes. My professor told me to use fgsea package, he also provided me with a data set from an article called “Genome-wide CRISPR Screens Reveal Host Factors Critical for SARS-CoV-2 Infection” (https://www.sciencedirect.com/science/article/pii/S0092867420313921#app2).
Data set: Table S1. Vero-E6 SARS-CoV-2 Genome-wide CRISPR Screens, Related to Figure 1.
This data set provided me with z-scores and gene names and gene ids. I have been working on trying to figure out how to use the package but keep running into, many errors. What I have done so far was create a data frame of my genes names, gene id and z-scores.
The example above uses my_pathways pathways <- reactomePathways(gene_id). But I get an error that says:
Error in .testForValidKeys(x, keys, keytype, fks) : None of the keys entered are valid keys for 'ENTREZID'. Please use the keys method to see a listing of valid arguments.
I was wondering if you have any advice for me. Im concluding that the data set I was provided with is not the correct data set in order to do this analysis.
Thanks,
Sal
Hi Sal,
the error is indicating that Entrez IDs are required and valid IDs were not found; please ensure that your gene IDs are Entrez IDs. Converting between different formats and identifiers is a common bioinformatics task. I have a blog post on converting between different gene identifiers that may be useful https://davetang.org/muse/2013/11/25/thoughts-converting-gene-identifiers/.
Cheers,
Dave